
The mycobacterial Lsr2 story is a curious one. Lsr2 showed up on the scene as an antigen from Mycobacterium leprae back in an Infection and Immunity paper published in 19911. Some work was done to sequence the antigen’s gene and the efforts of the 90’s revolved around leprosy and the immune response. Not much else was done for a while. If you poke around with BLAST, you will discover that lsr2 is well conserved in mycobacteria and actinomycetes, thus it is potentially interesting outside the realm of leprosy. Around 2006 to 2008, it was found that if you delete lsr2 in M. smegmatis it disrupts the ability to form biofilms2, changes the colony morphology from rough to smooth2 and results in a hypermotile phenotype3. Around the same time, publications came out that Lsr2 was involved in transcriptional regulation of multi-drug tolerance and antibiotic-induced responses4, as well as protecting against reactive oxygen intermediates5, in M. tuberculosis! This was intriguing, but the underlying molecular mechanisms still remained unknown.
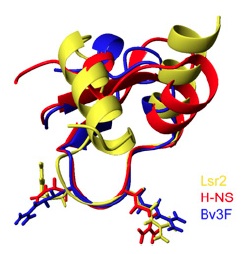
It is at this point, around 2008, that Jun Liu’s Lab (University of Toronto) started to shed some light on the mechanistic basis of Lsr2. They were able to show that M. tuberculosis Lsr2 is able to bridge distant segments of DNA6, much like the histone-like nuleoid structuring protein H-NS in E. coli. They could even complement an E. coli H-NS defect using M. tuberculosis Lsr27, suggesting that Lsr2 is an analogue of H-NS. This was exciting stuff, since Lsr2 and H-NS have no sequence homology and based on structural comparisons, the connection would not have likely been made. In their most recent articles (Gordon et al., 2010 and 2011), they used ChIP-on-chip analysis to perform high-resolution, genome-wide mapping of Lsr2 binding sites revealing that Lsr2 binds many genes involved in M. tuberculosis virulence and immunogenicity, including the ESX secretion system and PE/PPE genes. They also determined the tertiary structure of the Lsr2 DNA-binding domain, showing that the Arg97-Gly98-Arg99 residues are inserted into the minor groove of DNA at AT-rich regions. This is similar to the AT-hook motif in mammalian non-histone chromatin protein HMGA, which also binds AT-rich DNA and is involved in chromatin structure and gene regulation. Other residues in Lsr2 interact with the sugar-phosphate backbone on either edge of the minor groove and increase binding affinity. They then went on to determine the structural basis for recognition of AT-rich DNA by other unrelated silencing proteins, namely H-NS and Bv3F. In their 2011 paper,they show that it is the width of the minor grove that determines the binding preference of H-NS and Lsr2. The figure above (Fig. 3F from Gordon et al. 2011) shows the superimposed structures of Salmonella H-NS (red), Burkholderia Bv3F (blue) and Mycobacterium Lsr2 (yellow). You can see that the loop consisting of the conserved “T/SXQ/RGRXPA” motif of these proteins adopts nearly identical conformations. This is an excellent example of convergent evolution and definitely some Good Reads!
- Sela, S. (1991) Identification of Mycobacterium leprae antigens from a cosmid library: characterization of a 15-Kilodalton antigen that is recognized by both the humoral and cellular immune systems in leprosy patients. Infection and Immunity 59: 4117-4124.
- Chen, J.M., et al. (2006) Roles of Lsr2 in colony morphology and biofilm formation of Mycobacterium smegmatis. J. of Bacteriology 188: 633-641.
- Arora, K., et al. (2008) Inactivation of lsr2 results in a hypermotile phenotype in Mycobacterium smegmatis. J. of Bacteriology 190: 4291-4300.
- Colangeli, R., et al. (2007) Transcriptional regulation of multi-drug tolerance and antibiotic-induced responses by the histone-like protein Lsr2 in M. tuberculosis. PLoS Pathogens 3: 780-793.
- Colangeli, R., et al. (2009) The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. PNAS 106:4414-4418.
- Chen, J.M., et al. (2008) Lsr2 of Mycobacterium tuberculosis is a DNA-bridging protein. Nucleic Acids Research 26:2123-2135.
- Gordon, B.R., et al. (2008) Lsr2 of Mycobacterium represents a novel class of H-NS-like proteins. J. of Bacteriology 190:7052-7059.
At the level of gene regulation, phage λ is probably the best-understood organism on Earth. Yet, every time I think we have most of this phage figured out and there is not much else to learn, I read yet another interesting and insightful article about λ.
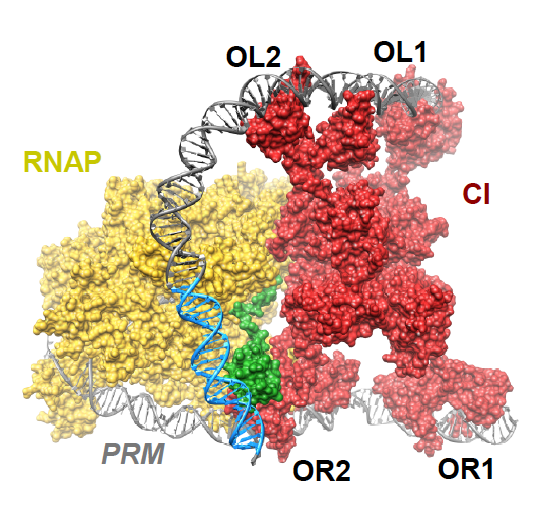
For those who are not familiar with phage λ genetic circuitry or don’t remember it from genetics class, here is a primer on the basics: The genetic switch controls whether an infecting phage undergoes lytic or lysogenic growth and in phage λ this is centrally controlled by whether or not the cI repressor gene is expressed. The default decision is to undergo lytic growth whereby the phage replicates within the host cell and lyses the cell, releasing progeny, unless repressor is made. CI is expressed from the PRM promoter and there is a divergent, lytic promoter, PR, which drives expression of cro. Cro antagonizes CI and drives lytic growth. These two promoters are both controlled by the operator OR, which is actually three operator sites (OR1, OR2, OR3). CI binds cooperatively to OR1 and OR2, shutting down expression from PR and thus lytic growth. It also activates its own expression at PRM. When concentrations of CI in the cell are sufficient it binds to OR3 and blocks its own transcription, thus maintaining a steady state of CI in the cell. Not only does CI bind to the OR sites cooperatively, but there are similar sites ~2.3 Kbp away at the OL operator that also binds CI. Further cooperativity between CI bound at OR and OL leads to DNA looping, further enhancing the activation and repression of the PRM and PR promoters, respectively. (If this is a bit much, there is a great minireview on λ genetics by Court, Oppenheim and Adhya1 that gives a more in depth explaination)
How DNA looping enhances repression of PR is pretty straight forward, but what has not been well understood is how the OL site increases CI activation of PRM. Cui et al. provide support for the “UP contact model” in which OL-OR looping brings an UP element for the PL promoter close enough to PRM so that it can be contacted by the C-terminal domain of the α-subunit of an RNA polymerase bound at PRM. They show that activation requires the UP element, an intact α C-terminal domain and CI-mediated DNA looping, thus confirming the model. Furthermore, they provide structural models supporting the feasibility of contact between the UP element and the α-subunit. The figure above (Fig. 4D in the article) shows an octamer of CI bound at two OR and two OL sites looping the DNA so that the α-subunit (green) of RNA polymerase (yellow) can contact the UP element (blue).
Enhancer elements are quite intriguing genetic elements that can activate transcription at a promoter located several kilobases away. They are widespread, existing in eukaryotes, prokaryotes and phage, yet our understanding of how enhancer-promoter complexes work at a mechanistic level still lacks many details. Cui et al. have uncovered an enhancer-like element in λ that both provides further insight into λ genetic circuitry and a potential model for enhancer elements. What wonders will be revealed about this 48.5 Kbp piece of DNA we call λ in the near future? I can’t wait to find out!
1. Court, D., et al. 2007. A new look at bacteriophage λ genetic networks. 189(2): 298-304.
Gregory Broussard
Revision: 4/19/12
(Please use, change and disseminate freely!)
- Design primers according to the Quickchange SDM protocol.
- Isolate plasmid DNA from a dam+ E. coli strain.
- Run the SDM PCR:
- Use 125 ng of each primer
- Use ~25 ng of plasmid template DNA
- Use 1 μl (2.5 U/μl) of Pfu Turbo DNA Polymerase
- 95 °C 30 sec.
- ——————
- 95 °C 30 sec.
- 55 °C 1 min. 12-18 cycles
- 68 °C 1 min./kb
- ——————
- 4 °C indef.
- Point mutations 12 cycles
- Single amino acid changes 16 cycles
- Multiple amino acid deletions or insertions 18 cycles
- Add 1 μl DpnI (10U/ul) directly to each PCR rxn. Mix in a 1.5 ml tube and incubate for 1 hr. at 37 °C. (This will digest the parental DNA; sometimes overnight digestion gives better results.)
- Transform 1 μl of each rxn into XL1-Blue or NEB5α cells and plate with selection. Incubate at 37 °C, overnight.
Notes:
- Do a control for DpnI activity:
- Do control PCR w/o primers
- Digest w/ DpnI, as with the true SDM rxn.
- Transform and plate. There should be less colonies on the control (0-2 cfu is ideal).
- Use 25-50 ng of template DNA in the PCR.
- Math for primer conc.:
(125 ng/(330 X number of bp)) X 1000 = number of pmole primer for 125 ng
primer stocks are 10 μM = 10 pmole/μl
So, for example add 0.9 μl of 10 μM primer for 9 pmoles
Gregory Broussard
Revised: 8/1/07
(Please use, change and disseminate freely!)
- When receiving a new lyophilized primer start by making a 100 µM stock. Multiply the number of nmoles printed on the tube by 10. This is the number of µl of TE to add to the tube to get a 100 µM stock. Make this stock directly in the tube containing the primer.
- Make a 10 µM working stock by making a 1:10 dilution of the 100 µM stock. Use this for your PCR reactions
Gregory Broussard
Revision: 1/25/11
(Please use, change and disseminate freely!)
Reaction:
3.5µL diH2O
40µL PCR Product
5µL T4 PNK Buffer (10X) (note 1 – use T4 DNA ligase buffer instead)
0.5µL ATP (100 mM) (note 1 – use T4 DNA ligase buffer instead)
1µL T4 PNK
———————–
50µL Total Vol.
Incubate at 37°C for 30 min.
Heat inactivate by heating at 65°C for 20 min.
Notes:
- 1X T4 DNA Ligase Reaction Buffer contains 1 mM ATP and can be substituted in non-radioactive phosphorylations (T4 PNK exhibits 100% activity in this buffer)
- Fresh buffer is required for optimal activity (in older buffers, loss of DDT due to oxidation lowers activity)
- Often, a kinase reaction is followed by a ligation reaction. In such cases, the T4 PNK reaction is performed in ligase buffer at 37°C for 30 min. The product of this reaction can be used directly in the ligation reaction without a buffer change or heat inactivation UNLESS there is a need to keep other DNA fragments dephosphorylated during ligation. When this is desirable, PNK should be heat inactivated prior to ligation.
Gregory W. Broussard
Revised: 9/10/10
(Please use, change and disseminate freely!)
- Place 1 mL culture in a 1.5 mL tube
- Centrifuge at 5,000 X g for 5 min.
- Resuspend pellet in 500 µL TE
- Place at 95°C for 20 min.
- Place on ice for ~1 min. VORTEX on high setting!!!
- Centrifuge to pellet, 13,000 rpm for 1 min.
- Save supernatant in a 1.5 mL tube
- Use 10 µL of this as the template in PCR reactions
Gregory W. Broussard
Revised: 8/3/07
(Please use, change and disseminate freely!)
- Use primers at a concentration of 10 μM
- Using DMSO at 5% may help the reaction, especially with DNA of high GC content (ex: Mycobacterium chromosomal DNA). For M. smegmatis and mycobacteriophages, I now always use 5% DMSO.
- 50 μl total volume
- This protocol is for Pfu DNA Polymerase (Stratagene)
- For mycobacteriophages as template, use 1μl of lysate in place of template DNA.
- PCR Rxn Mix:
| Components | Stock Concentration | Final reaction Concentration |
50 ml reactiona |
| M. smegmatis DNA | 5 – 25 ng/μl | 0.1 – 0.5 ng/μl |
1 μl |
| Primer A | 10 μM | 0.25 μM |
1.25 μl |
| Primer B | 10 μM | 0.25 μM |
1.25 μl |
| dNTP mixture | 10 mM | 0.2 mM |
1 μl |
| 10X buffer | 10X | 1X |
5 μl |
| DMSOb | 100% | 5% |
2.5 μl OR 0 μl |
| Pfu | 2.5 U/μl | 2.5 U |
1 μl |
| dH2O | — | — |
37 μl OR 39 μl |
- Annealing temperature should be determined by looking at the melting temperature (Tm) for both primers. It is recommended to choose an annealing temperature 2 °C lower that the lowest Tm.
- Extension time for small PCR products (< 1000 bp) should be 1 min. 30 sec.
- PCR:
| Cycle function | Number of cycles | Temperature | Time length |
| Initial denaturation | 1 | 95°C | 5 min |
| Denaturation | 95°C | 30 sec | |
| Annealinga | 30 | —— | 30 sec |
| Extension | 72°C | 1 min/kb + 30 sec | |
| Final Extension | 1 | 72°C | 7 min |
| Hold | 1 | 4°C | Indefinite |
- Run 5 μl of the PCR product on a gel.
Gregory Broussard
Revision: 02/24/11
(Please use, change and disseminate freely!)
- Use a culture of M. smeg grown to an O.D.600 of 0.8-1.0.
- Dilute phage lysate to 1010 pfu/ml
- Ex: (1.6 x 1011pfu/ml) X = (2.0 x 1010pfu/ml)(1ml)
- X = 125 µl lyate into 937.5 µl Phage buffer/CaCl2
- Plate 100 µl diluted lysate onto plates for a total of 2 X 109 pfu/plate
- Make 10-1 to 10-7 dilutions of M. smeg strains
- Plate 100 µl of diluted M. smeg onto phage seeded plates and plates not seeded with phage (I usually plate 10-4 to 10-7 dilutions).
- Incubate at 37°C for ~4 days (wrapped in parafilm or bagged).
- Count cfu’s and divide cfu’s on phage seeded plates by those without phage and multiply times 100 to get percent lysogeny.
Gregory Broussard
Revised: 8/1/07
(Please use, change and disseminate freely!)
- Start by growing a 5 ml culture to mid-log phase.
- In a well-labeled screw capped tube add 500 µl culture to 500 µl 40% glycerol.
- Mix well by inverting the tube about six times or by vortexing.
- Place the tube in the -70 ºC freezer for indefinite storage.
Gregory W. Broussard
Revised: 04/21/10
(Please use, change and disseminate freely!)
- Pick Mycobacterium colonies carrying your plasmid of choice into 20 µl of ice-cold 10% glycerol.
- Vortex tube briefly before placing on ice for 10 min.
- Add Myco-10% glycerol mix to electrocompetent E. coli cells
- Electroporate using the following parameters: 200 Ω, 2500 V, 25 µF
- Recover in 900 µl SOC broth at 37 °C for 1 hr.
- Plate on LB plates with selective antibiotic.
- Incubate at 37 °C overnight.
Gregory W. Broussard
Revised: 07/13/11
(Please use, change and disseminate freely!)
- 15 µl ligation final volume
- Vector mass should be 100 ng
- For vector and insert fragments of about the same size use 1:1 molar ratio
- If insert is much smaller (<1000bp) use 1:3 (vector:insert) ratio
- For blunt end ligations use a 1:5 (vector:insert) ratio
1. Use the following equation to determine the volumes of insert and vector:
bp insert bp vector
———– = ———–
X ng insert 100 ng vector
- “X” equals the ng of insert for a 1:1 ratio
- for a 1:3 ratio multiply X times 3
- with this information and previous measurements of concentration, determine volumes of each to add to the ligation mix.
2. Set up ligation mix:
(total vol. 15 µl)
? µl diH2O
? µl vector (100 ng)
? µl insert (see formula)
1.5 µl ligase buffer
1.5 µl ATP (0.75 µl for blunt ends)
1 µl ligase
- If you are using the Fast-Link DNA ligase kit, incubate at RT for 1hour
- Manufacturer recommends shorter time, but longer is better here
- If one enzyme has a blunt end cut, you MUST use the amounts of ATP for blunt ligations (too much ATP can inhibit the ligation.)
3. Transform into your favorite bug!
4. You can store the ligation at 4 or -20 ºC.
At the beginning of my postdoctoral work in Dr. Graham Hafull’s Lab at the University of Pittsburgh, I isolated a mycobacteriophage from soil my wife collected from my home state of Louisiana. I called it Acadian, because it was isolated in the heart of Cajun (Acadian) country. The genome was subsequently sequenced and found to be the first member of the new subcluster of mycobacteriophages, subcluster B5. It is submitted to GenBank accession no. JN699007.1. A wonderful compilation of information on this and other mycobacteriophages can be found at phagesdb.org.
(This protocol works, I just don’t know the transformation efficiencies at this time)
Gregory W. Broussard
Revised: 5/31/12
(Please use, change and disseminate freely!)
This protocol is for a 100 mL culture and will yield ~40, 100 mL aliquots.
- Inoculate two 5 mL cultures of E. coli NEB5alpha cells in LB broth and grow overnight.
- Subculture into a 500 mL flasks (nonbaffled) by adding 3 ml of one of the culture from #1 into 100 mL of the same media. Check O.D.600 and incubate shaking at 37° C for 2-4 hours.
- When cells reach an OD600 of ~0.5, transfer the cells to two 50 mL conical tubes. Incubate the tubes on ice for 30 min to 1 hr.
- Centrifuge the cells at 5000 rpm for 10 min and discard the supernatant.
- Wash the cells in 1/2 vol. (25 mL) 10% glycerol and centrifuge as above.
- Wash the cells in 1/4 vol. (12.5 mL) 10% glycerol and centrifuge as above.
- Wash the cells in 1/8 vol. (6.25 mL) 10% glycerol and centrifuge as above.
- Suspend the cells in 2 mL 10% glycerol.
- Separate into 100 mL aliquots in microcentrifuge tubes.
- Flash freeze on dry ice and store at -80° C or use immediately.
Gregory W. Broussard
Revised: 01/20/09
(Please use, change and disseminate freely!)
This procedure is used for M. smegmatis and M. tuberculosis electrocompetent cell preparations. M. smeg cells should be kept cold by using ice-cold glycerol and centrifuging at 4° C. M. tb cells and glycerol should be kept warm and centrifugations should be performed at RT. These guidelines will improve transformation efficiency. This protocol is for a 50 mL culture and will yield ~20, 100 µL aliquots.
- Inoculate a 3 mL culture of M. smeg mc2 155 in 7H9 broth (containing ADC, CB, CHX, Tw) and grow until saturation (~2 days).
- Subculture into 50 mL of the same media in a 250 mL flask (nonbaffled) to a final OD600 = 0.020 and grow shaking at 37° C overnight.
- On the following day, when cells reach an OD600 of 0.8-1.0, transfer the cells to 50 mL conical tubes. For M. smeg, incubate the tubes on ice for 30 min. to 2 hrs.
- Centrifuge the cells at 5000 rpm for 10 min and discard the supernatant.
- Wash the cells in 1/2 vol. (25 mL) 10% glycerol and centrifuge as above.
- Wash the cells in 1/4 vol. (12.5 mL) 10% glycerol and centrifuge as above.
- Wash the cells in 1/8 vol. (6.25 mL) 10% glycerol and centrifuge as above.
- Suspend the cells in 3 mL 10% glycerol.
- Separate into 100 µL aliquots in microcentrifuge tubes.
- Freeze on dry ice and store at -80° C or use immediately.
Notes:
- Using M. tb immediately will improve efficiency.
- If arching occurs after thawing cells, do two mini washes in 1 mL 10% glycerol and resuspend in 100 µL for electroporation.
Gregory W. Broussard
Revised: 10/22/07
(Please use, change and disseminate freely!)
This transformation procedure assumes the use of a BioRad pulse-controller electroporation unit. If using fresh M. tb cells, without freezing, the cells are not incubated on ice after adding DNA.
- Thaw tubes of cells on ice for ~10 min.
- Add DNA (1-50 ng, 100ng for linear targeting substrate DNA) to cells and mix gently. Incubate for 10 min. (on ice for M. smeg).
- Transfer cells to a cuvet (pre-chilled for M. smeg).
- Wipe all moisture off of the cuvet and place in electroporator holder. Electroporate with the pulse controller set to 2.5 kV, 1000 Ω, 25 μF. Check time constant, it should be between 18-22. If the cells arc, repeat the transformation (possibly do further washes with 10% glycerol).
- For recovery, 1 mL 7H9 broth (containing ADC and Tw, and oleic acid if for M. tb) is added to the cells and cells are transferred to a test tube and incubated at 37° C. For plasmid DNA in M. smeg, recover for 2 hrs., in M. tb recover overnight. For transformations with linear targeting substrate DNA recover for 4hrs. in M. smeg and for 72 hrs. in M. tb.
- Plate on selective media and incubate at 37° C.
Gregory Broussard
Revised: 11/24/08
(Please use, change and disseminate freely!)
Disclaimer: This protocol DOES NOT give efficient electroporations. It will give you 10-30 colonies with 1 µl of prepped plasmid. ONLY use with supercoiled DNA when you need to get stuff done TODAY!!!
- Centrifuge 1.5 ml culture in a microfuge tube at 5,000 X g for 1 minute.
- Resuspend pellet in 1 ml cold 10% glycerol. (keep on ice when not centrifuging) Centrifuge again.
- Repeat step #2 twice more for a total of three washes with cold 10% glycerol.
- After the final wash suspend cells in 100 µl cold 10% glycerol and place on ice.
- Add 1 µl or desired amount of plasmid DNA and sit on ice for 10 min.
- Electroporate at 2.5kV, 1000 Ω, 25 µF.
- Add 1 ml 7H9/ADC/Tw to cells.
- Recover for 2 hrs. at 37 ºC, shaking.
- Plate with appropriate antibiotics.
Gregory Broussard
Revised: 09/22/08
(Please use, change and disseminate freely!)
This is for when you need to do a transformation TODAY and all you have is a fresh culture of your bacteria, but NO COMPETENT CELLS…and you just need one colony harboring your favorite plasmid. This is NOT for transformation efficient cells, but can save you a day’s time!!!
- Pellet 1 ml of cell culture
- Add 100 µl of 50 mM COLD CaCl2
- Add 1 µl plasmid DNA
- Incubate on ice for 30 min
- Heat-shock at 42ºC for 45 seconds
- Immediately place tube on ice for 2 min
- Add 450 µl TSB and gently mix
- Incubate at 37ºC for 1 hour (*not any longer)
- Plate on LB agar containing the correct antibiotics
- It is recommended to plate 100 µl on one plate and plate the rest on another plate to avoid having a lawn
- Incubate overnight @ 37ºC
Gregory Broussard
Revised: 07/20/07
(Please use, change and disseminate freely!)
Transformation of chemically competent E. coli:
- Thaw cells on ice for about 10 min, one tube of 50µl cells per transformation
- Add DNA (1-50 ng plasmid) and mix gently by tapping
- Incubate on ice for 30 min
- Heat-shock at 42ºC for 45 seconds
- Immediately place tube on ice for 2 min
- Add 450 µl TSB and gently mix
- Incubate at 37ºC for 1 hour (*not any longer)
- Plate on LB agar containing the correct antibiotics
- It is recommended to plate 100 µl on one plate and plate the rest on another plate to avoid having a lawn if transformation was very efficient
- Incubate overnight @ 37ºC
Transformation of electrocompetent E. coli:
- Thaw cells on ice for about 10 min, one tube of 50 µl cells per transformation
- Place cuvettes on ice, one per transformation
- Add DNA (1-50 ng plasmid) and mix gently by tapping
- Incubate on ice for 10 min
- Pipet into chilled cuvette
- Wipe ice/water off the cuvette and tap to remove bubbles
- Place in electroporator holder
- Electroporate: Set to 2.5 kV, 200 Ω, 25 ºF
- Push both buttons simultaneously and hold until beeping sound
- If it arcs you will hear a popping sound, the cells are likely dead and this transformation must be repeated
- Add 1 ml TSB to electroporated cells
- You can add the media directly to the cuvette and then gently pipet it back out into a test tube
- Incubate at 37 ºC for 1 hour (*not any longer)
- Plate on LB agar containing the correct antibiotics
- It is recommended to plate 50 µl on one plate and plate the rest on another plate to avoid having a lawn (we want single colonies)
- Incubate overnight @ 37 ºC
Gregory W. Broussard
Revised: 7/10/08
(Please use, change and disseminate freely!)
- Inoculate 5 mL M7H9/ADC/CHX/CB/Tween80 with your strain of M. marinum. Incubate at 30°C shaking until turbid.
- Inoculate from 50-100 mls broth (in 250-500 ml non-baffled flask) with the 1 ml culture. Place culture shaking at 30°C until O.D.600 = ~0.6-0.9.
- Transfer to 50 ml conical tubes and centrifuge 5,000 rpm for 10 min.
- Resuspend the pelleted cells in 1/10th original volume of M7H9/ADC. (5mls for 50 mls original vol.)
- Add the same volume of 40% glycerol and mix.
- Aliquot 200µl per 1.5ml tube.
- Freeze at -80°C.
- The following day streak one tube onto a M7H10 plate to check for purity.
Gregory W. Broussard
Revised: 2/27/08
(Please use, change and disseminate freely!)
- Digest plasmid DNA to recover insert and vector fragments.
Ex: 23µL diH2O
20µL DNA
5µL NEB2
1µL HindIII
1µL NdeI
50µL, 37°C, 4hrs.
- Heat inactivate Restriction Enzymes if possible.
Ex: 65°C for 20min.
Or, remove enzymes by other method.
- Add BSA (if not already added), and dNTPs (100µM).
Ex: Add 0.5 µL dNTPs (10mM) and 0.5 µL BSA (10mg/ml)
- Add 1µL T4 DNA Polymerase (or 1 unit per µg DNA). Incubate for 15 min. at 12°C. (NOT ANY LONGER)
- Stop the reaction by adding EDTA to a final concentration of 10mM and heating to 75°C for 20 min.
Ex: Add 1µL 0.5M EDTA to the above reaction.
- Gel purify the fragment of choice.
Caution: Elevated temperatures, excessive amounts of enzyme, failure to supplement with dNTPs or long reaction times may result in recessed ends due to the 3′ to 5′ exonuclease activity of the T4 DNA Polymerase enzyme!!!
Gregory Broussard
Revision: 07/29/11
Reagents:
Saturated ammonium sulfate: Bring 80 g ammonium sulfate to 100 ml in dH2O, and stir for > 2 hrs on stir plate. Allow the undissolved crystals to settle. Remove solution, leaving behind undissolved crystals, and place in 50 ml conical tubes.
95% EtOH
70% EtOH
Buffer-equlibrated phenol
Phenol:chloroform:isoamyl alcohol (25:24:1)
Chloroform
RNAse A (10mg/mL)
DNase I (2,000 U/ml)
TE
7H9+agarose plates
MBTagarose
Methods:
- Prepare plates of 7H9 (4.7g), agarose (15g), 10 ml 0.1 M CaCl2, 2-3 drops antibubble and 12.5 ml 40% glycerol in 900ml H20; add ADC, CHX, and CB after autoclaving. (The use of agarose instead of agar remedies a problem of co-purification of a substance from the agar that inhibits some restriction enzymes and possibly other types of enzymes). Also, prepare MBTagarose by adding agarose in the place of agar in the MBTA recipe.
- Plate dilutions of phage on normal 7H10 plates to determine the dilution that gives you “webbed” plates.
- Plate for “webbed” plates of your phage: use 7H9+agarose plates and MBTagarose. (4 plates is usually appropriate)
- Prepare a lysate and filter (0.22 mm).
- Add an equal volume of saturated ammonium sulfate, invert tube to mix gently, and incubate on ice 2 hrs.
- Pellet at 3500 x g for 10 min. at 4°C in a 50 ml conical tube. Mark the tube where the pellet is expected. Pellet this twice, but do not remove the supernatant after the first spin!!! The first time, the phage are in “chunks” of glassy pellet that you can see when inverting the tube gently. The second time, the pellet is more visible. Immediately remove the supernatant the second you take it out of the centrifuge. (If no pellet is seen, try centrifuging for 30 min.)
- Resuspend phage in 500 ml Phage buffer.
- During the above centrifugations, soak ~4” of dialysis tubing (15,000 MWCO) in 1L diH2O (for ~10 min.)
- Dialyze your 500 ml phage sample at 4°C with stirring, twice in phage buffer: once overnight and once for two hours (the order does not matter).
- Transfer sample to a 1.5 ml tube (unless volume is >700ml, then use 2 tubes).
- Check the volume and add the appropriate amount of 10X DNase I reaction buffer (final conc. 1X).
- Add 1ml of DNase I (2,000 U/ml; total of 2U) and X ml RNase A (10mg/ml; final conc. 100mg/ml) and incubate at 37°C for 1 hr. (DNase I will remove trace amounts of M. smeg DNA, the RNase A removes precipitated M. smeg ribosomes)
- Add an equal volume of buffer equilibrated phenol, mix until milky white, centrifuge at 13K rpm at RT for 5 min.
- Save aqueous phase, repeat phenol extraction until white interface is gone (~3-5 times total). Also, back-extract with 600 ml TE.
- Add equal volume of phenol:chloroform:isoamyl alcohol (25:24:1), mix until milky white, centrifuge as above.
- Save aqueous phase and add equal volume of chloroform, mix and centrifuge as above.
- Save aqueous phase, add 1/10 vol. 3M sodium acetate and 3 vol. 95% ethanol. Mix gently and well by inverting tube repeatedly until DNA precipitates and forms a nice “cotton ball”. (Do Not Vortex)
- Freeze on dry ice. Thaw on top of ice. Centrifuge at 14K rpm at 4 °C for 30 min.
- Remove ethanol leaving pelleted DNA in tube. Add 1 ml 70% ethanol and mix gently by inverting tube 6-8 times (pellet should float around in ethanol). Centrifuge at 13K rpm at RT for 1 min.
- Remove ethanol, pipette out any droplets, and air dry (~20 min.). Don’t let the DNA over dry or it will be hard to dissolve later, but do make sure all the ethanol has evaporated.
- Dissolve DNA in TE at 42°C for ~10 min. or until dissolved. Pipette gently to resuspend DNA. (Start with 50 ml TE, then add more if needed. It may be a good idea to add no more than 100 ml TE and leave it at RT overnight, then add more TE if needed.) Store at 4 °C.
- Measure the concentration of DNA on the nanodrop, measure undiluted DNA twice and 1:10 dilution once, make sure all readings are consistent.
Notes:
- This procedure will yield ~50-150 µg DNA
- Titers of dialyzed phage equal 1010 to 1012 pfu/ml
- Transformation of M. smeg with 100 ng BPs DNA yielded 104 pfu/µg
- PCR on BPs DNA gave product with BPs specific primers and no product with M. smeg specific primers
- BPs DNA gave correct band patterns when digested with BamHI, ClaI, EcoRI, HaeIII, KasI, SacI, NotI
- Dialyzed phage treated with RNase A has NOT been used at this time for EM work
